Docking Adventures with Boltz-2 and Nanome AI
Predicting protein-ligand interactions can be a complex task, but tools like MARA are making it significantly easier to assist with and automate the preparation of configuration files, and visualize a system analytically.
In this tutorial, we explore a streamlined workflow for generating Boltz-2 YAML configuration files for docking experiments. Following a prediction we will have MARA prepare a workspace with our docking results, each colored by Predicted Aligned Error (pae) and predicted Local Distance Difference Test (plddt) from the resulting .npz files provided after a Boltz-2 prediction.
YAML Configuration
A dedicated workflow (Boltz Docking with Pocket Constraint) was designed to
- retrieve and prepare protein coordinate files, and
- to obtain ligands and convert directly to a SMILES string for docking experiments.
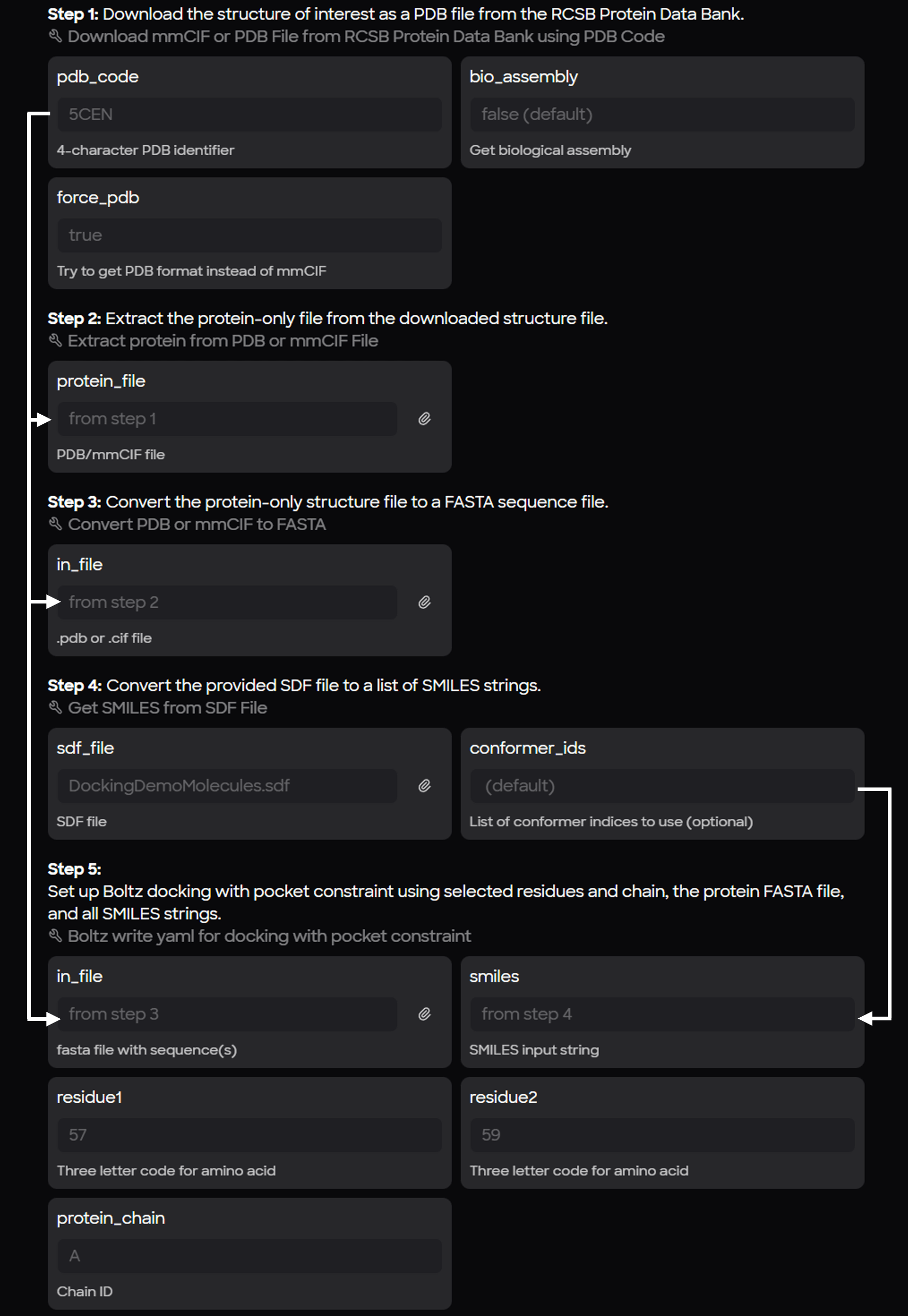
Primary steps of the workflow:
- Protein Retrieval: Downloading a protein (specifically in PDB format) from the RCSB.
- Cleaning: Removing water molecules from the crystal structure to create a protein-only file.
- Sequence Extraction: Converting the amino acid residues from the PDB into a FASTA file.
- Ligand Processing: Converting an SD file containing multiple small molecules into individual SMILES strings.
Multiple molecules in the SD file produces multiple yaml configuration files.
- Config Generation: Combining the protein sequence and SMILES string for each ligand into the final Boltz-2 configuration format.
Preparation Considerations: Residue Numbering
One of the most important aspects of setting up a Boltz-2 prediction for docking results is the residue numbering. Boltz-2 starts its numbering at one, regardless of the original numbering in the crystal structure.
For example, while the protein 5CEN actually starts at residue 117 in the crystal structure, the workflow identifies residues 57 and 59 (originally 174 and 176, respectively) for contact constraints because the predicted output from Boltz will be renumbered to begin from one. This ensures Boltz applies the proper constraints to Binder B from Chain A while completing a prediction.
Predicting from Scratch vs. Structural Inputs
While Boltz can be forced to employ a structural template and build off of existing coordinates, -template tag can be used with a path to a structural file, this workflow intentionally omits the protein PDB from the final Boltz input.
This forces a new prediction for each docking run by providing only the sequence and a ligand SMILES string. Researchers can then compare Boltz’s predicted protein fold against the known crystal structure to see how the software handles the docking across various iterations. An alignment within Nanome provides an RMSD value when comparing the folds from Boltz to the crystal structure.
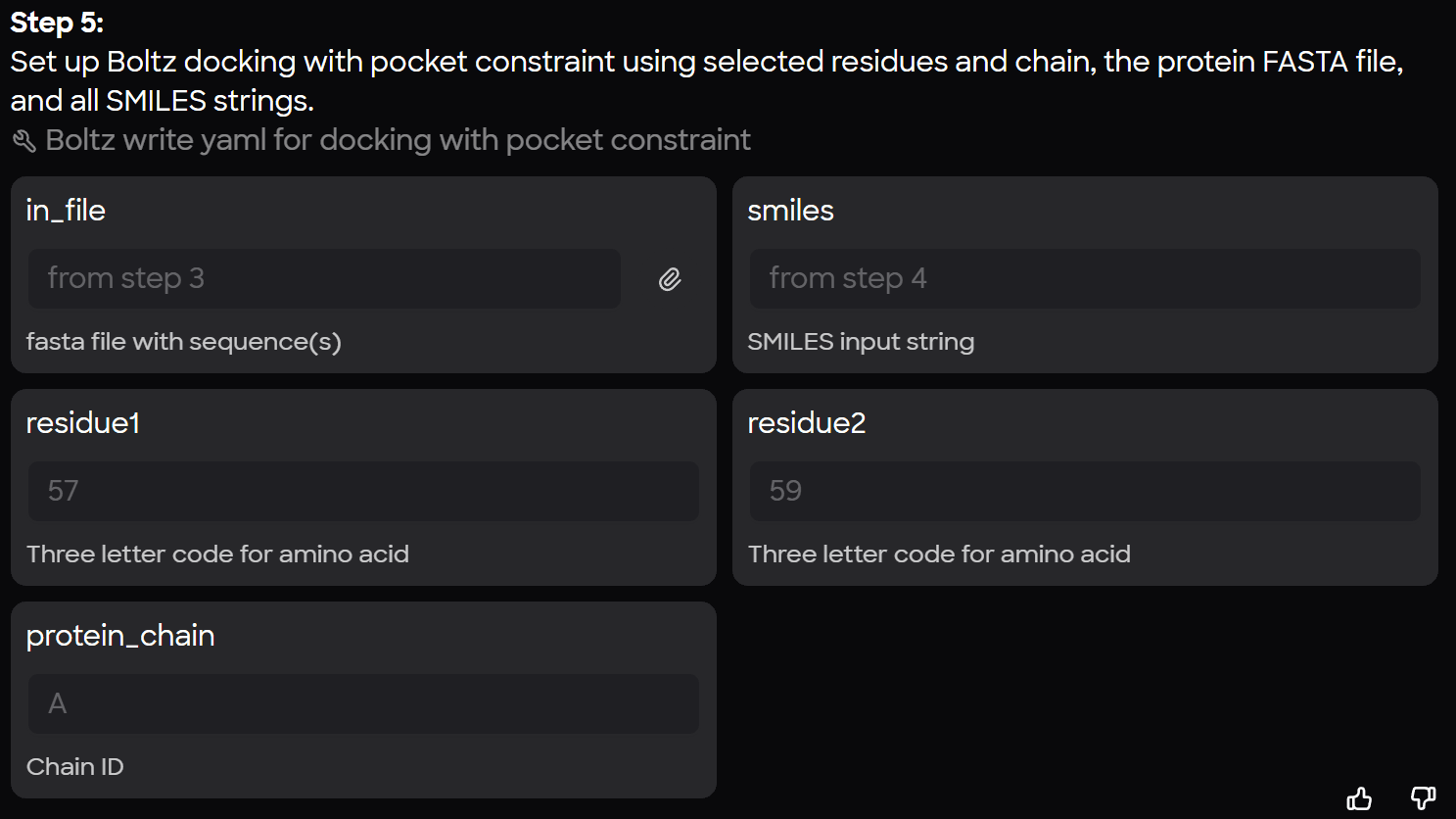
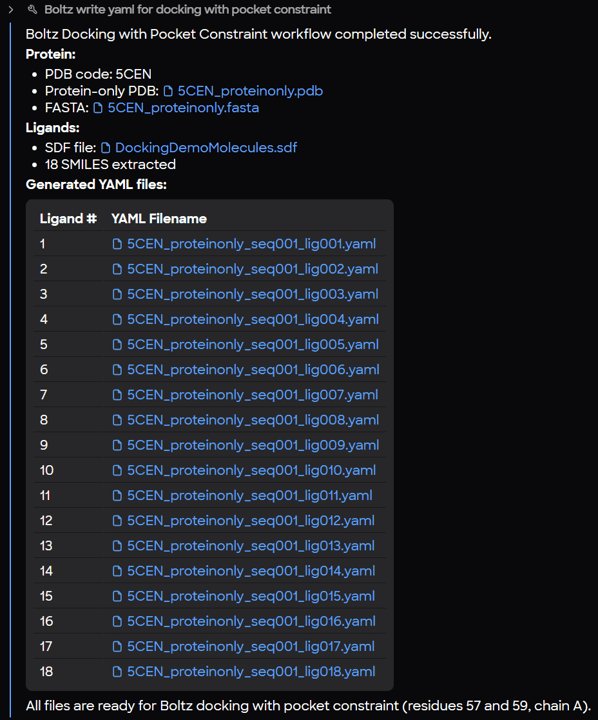
Create a Library of YAML Output Files from MARA
Once the workflow is executed, MARA generates a report confirming the creation of the input files. Each file contains:
- Protein Definition: Specifies the chain and the full amino acid sequence.
- Ligand Definition: Includes a unique ID and the specific SMILES string.
- Constraints: Defines the residues and chain from the protein that will interact with the ligand.
By passing a single protein and an SD file with multiple ligands into MARA, you can instantly generate dozens of YAML files, each ready for computation in Boltz-2.
Visualization of PAE and pLDDT Values per Structure
Understanding the results of a protein prediction or an affinity prediction requires understanding the system from many angles. MARA has built in tools to take a pdb output from Boltz and easily apply a color scheme derived from the pae and plddt analysis .npz files produced after a Boltz prediction.
In the prompt below (color each pdb by pae and plddt), 15 files were uploaded at once and MARA ran all the tools necessary to produce new files for each protein scaffold colored appropriately.
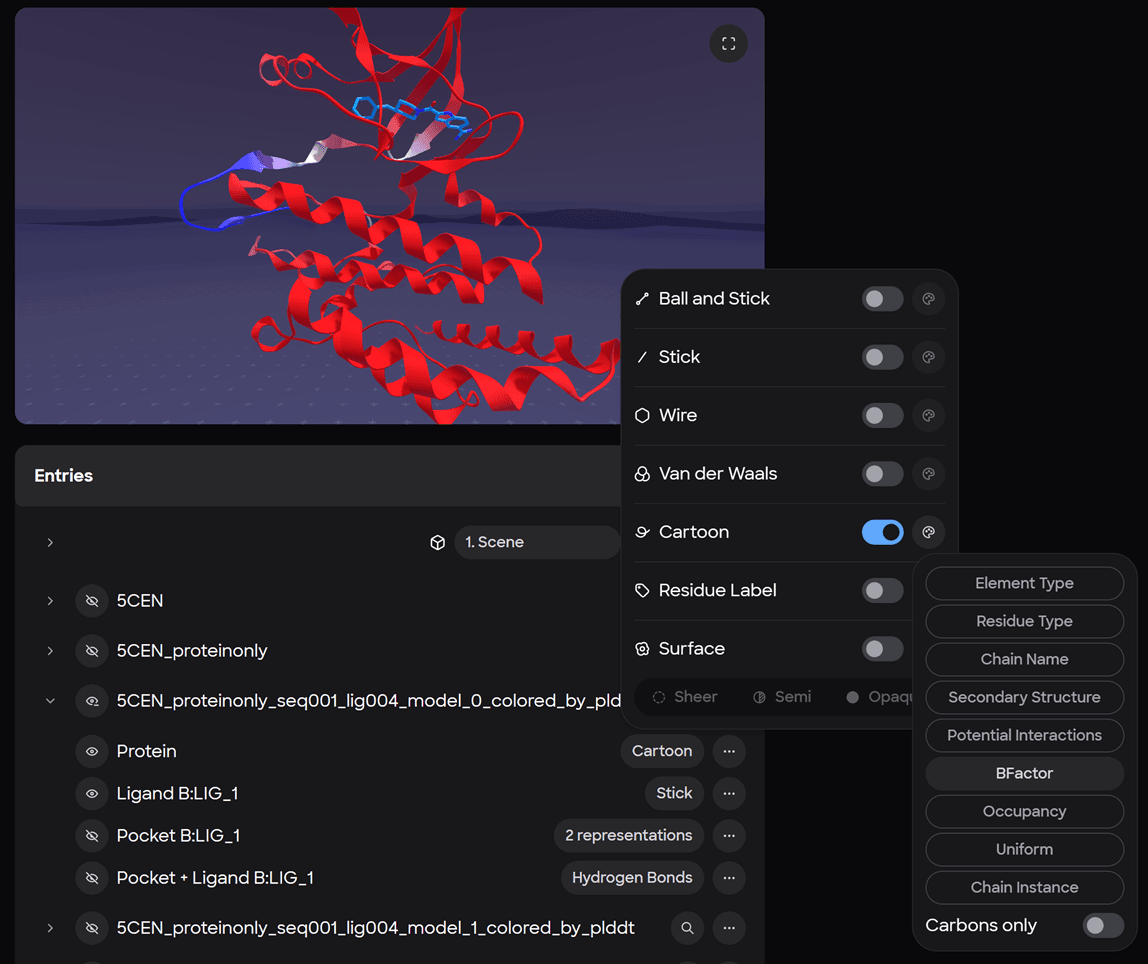
Visual Analysis with MARA Preview or in XR with Nanome V2
Asking MARA to create a new workspace, move all the pdbs, and color them by beta factor is all it takes for the workspace setup.

To make things easy, we wrote the pae and plddt scores to the beta factor column in the pdb file.

Clicking on a workspace id brings you directly to that workspace, where the “Preview” is waiting so you can ensure the workspace is set up properly before launching into XR. The Preview can be used in two modes, in the default mode users can interact with the entry list and make representation changes using the UI; fullscreen mode is also available to provide a more detailed view.

*Representations changes can also be made with MARA using natural language (and voice to text where supported) directly in a workspace allowing users to easily switch back and forth between between multiple coloring schemes
Multiple scenes containing targeted analytical visualizations can be prepared on the web ahead of an XR investigation. This allows users on the web to set a perspective for the scene so when they arrive in that scene with the augmented reality device they will already be in the action and not be required to find their protein of interest and set up everything again from scratch.
Concluding Remarks
The Nanome AI ecosystem acts as a single repository where all your files are at your fingertips. Comparing and contrasting multiple docking experiments with various ligands can be tedious when collecting the results from many different folders.
Employing MARA as a place where your protein sequences and molecules can live, and are easily retrievable; makes setting up a docking run easy for anyone on your team. As they will always have access to Workflows like the one in this blog and they can easily convert a pdb to fasta file and convert any sdf to SMILES. Tools that streamline coloring a prediction by analysis files make it that much easier to predict just one more structure.